
Genome-wide analyses of the past few years have revealed that a substantial portion of the genetic control of complex traits is exerted through the regulation of gene expression. Much of the genetic variation associated with complex traits falls outside the protein coding regions of genes. Mechanistic understanding of how this variation contributes to phenotype is lacking, but gene regulation is thought to play a major role. We develop and apply methods that fully harness gene regulation within complex trait association and prediction studies.
PrediXcan Across Populations
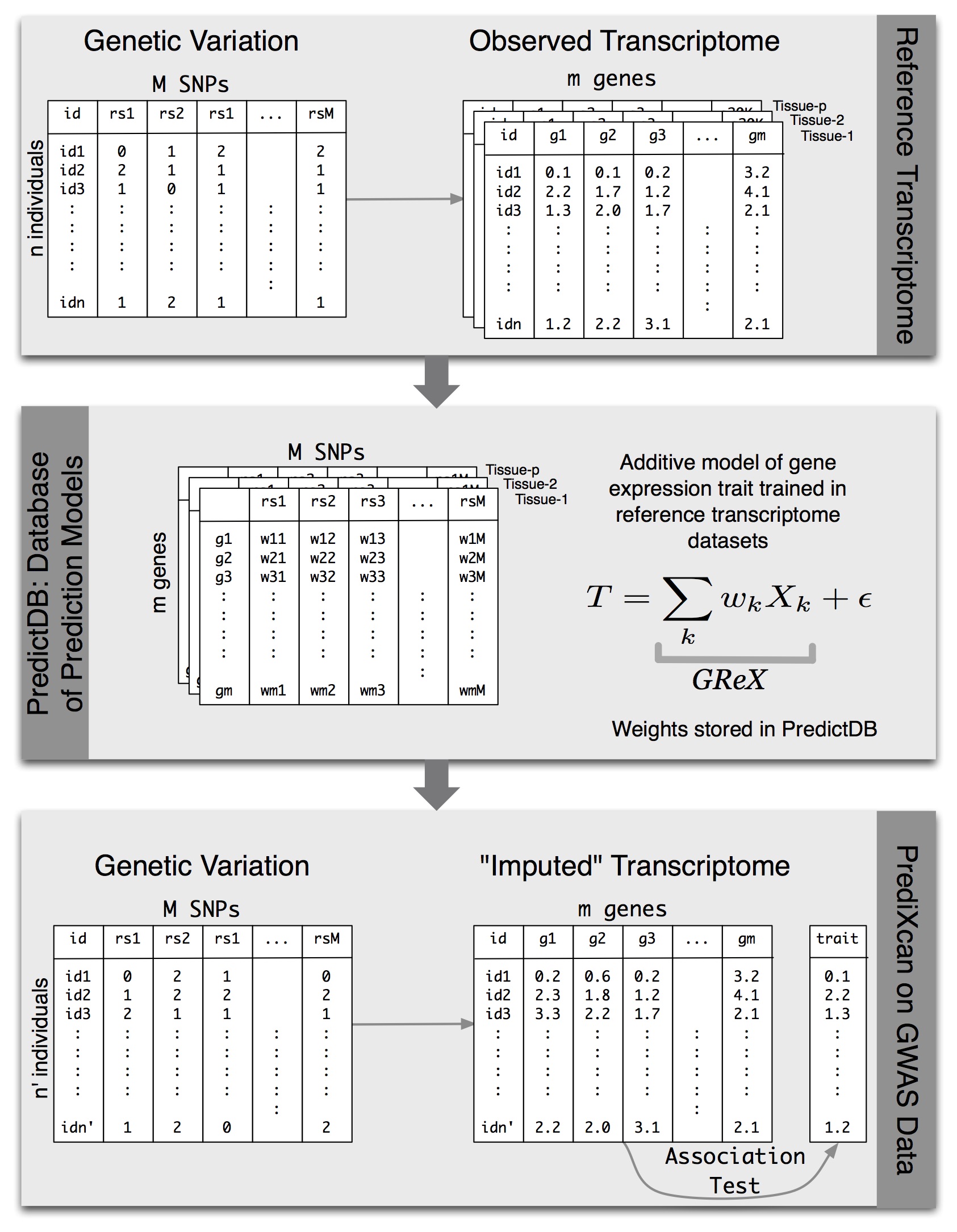
We have developed a gene-based association method called PrediXcan (pronounced “predict-scan”) that directly tests the molecular mechanisms through which genetic variation affects phenotype (published in Nature Genetics). The approach estimates the component of gene expression determined by an individual’s genetic profile and correlates the “imputed” gene expression with the phenotype under investigation to identify genes associated with the phenotype. The genetically regulated gene expression is estimated using whole-genome tissue-dependent prediction models trained with reference transcriptome datasets. The PrediXcan novel gene-based test is focused on the mechanism of gene regulation and comes with key information on direction of effect (i.e. whether high or low gene expression is associated with disease risk). Directional information can help to close in on potential drug targets.
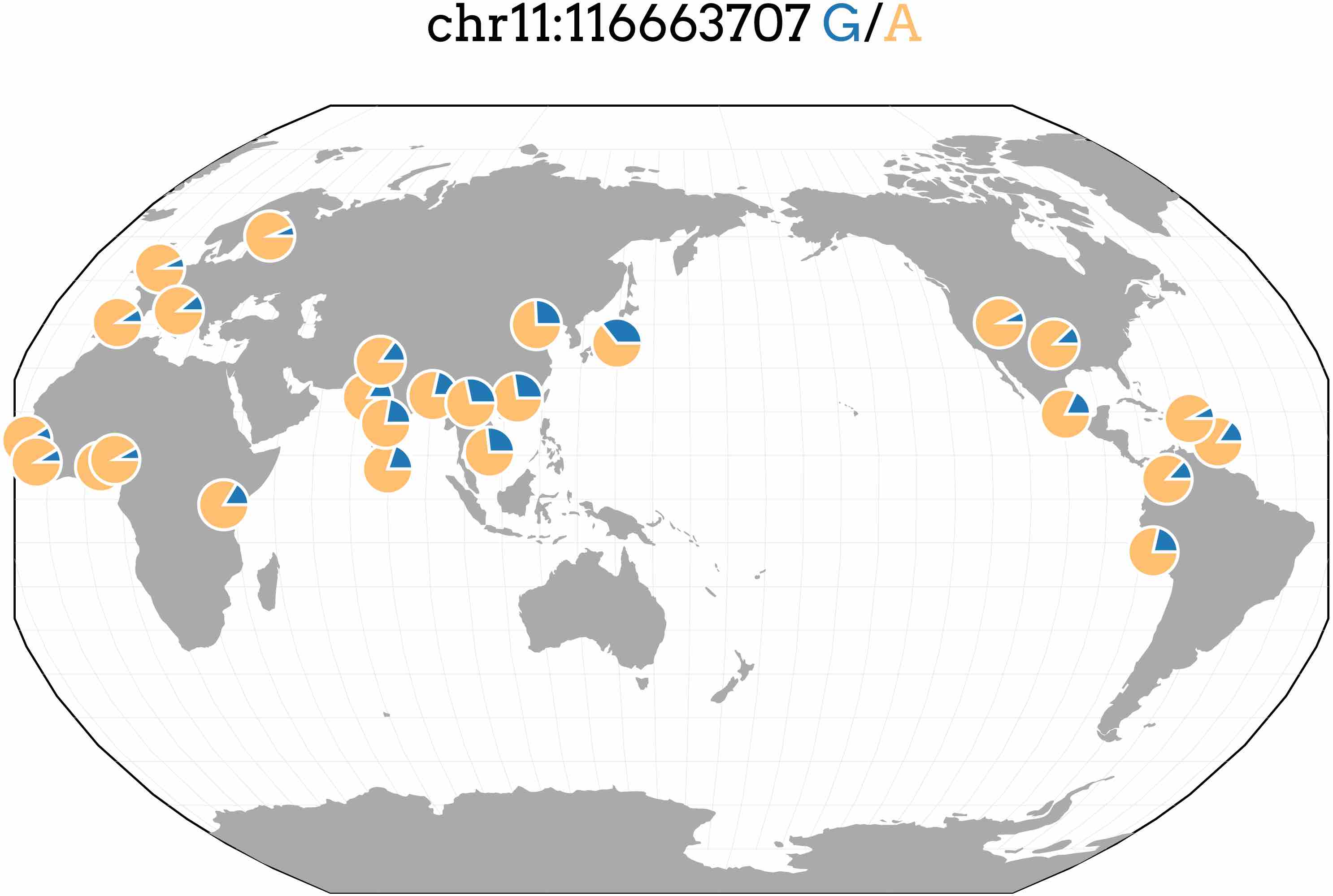
Most genome-wide association studies (GWAS) have been conducted in populations of European ancestry leading to a disparity in understanding the genetics of complex traits between populations. We performed GWAS and PrediXcan on four lipid traits in cohorts from Nigeria and the Philippines and compared them to the results of larger, predominantly European meta-analyses. The top SNP in the Filipino cohort associated with triglyceride levels (rs662799; P = 2.7 × 10−16). While this SNP is located directly upstream of previously known APOA5, we show it may also be involved in the regulation of BACE1 and SIDT2. rs662799 has a lower minor frequency in European and African populations than Asian populations (see below). Thus, it has a higher allelic impact in Asian populations, demonstrating how prediction models may vary in utility across populations. See our article in PeerJ for more details.
Genetic Architecture of Gene Expression Across Populations
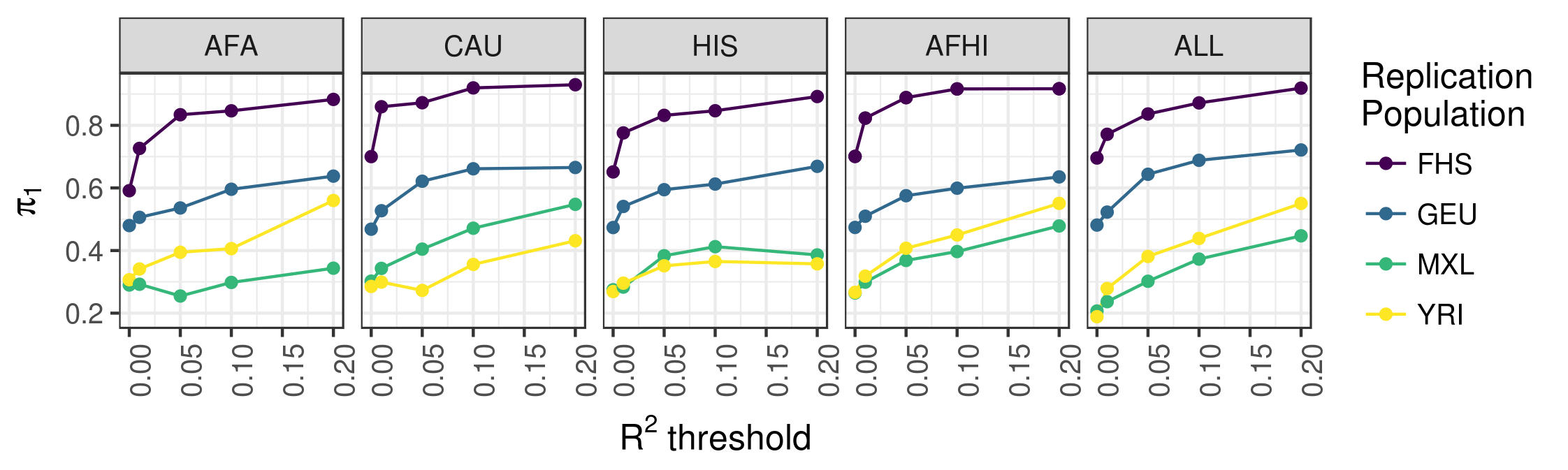
We used data from MESA to study the underlying genetic architecture of gene expression by optimizing gene expression prediction within and across diverse populations. After calculating the prediction performance, we found that many genes that were well predicted in one population are poorly predicted in another. We further show that a training set with ancestry similar to the test set resulted in better gene expression predictions, demonstrating the need to incorporate diverse populations in genomic studies. Our eQTL summary statistics and gene expression prediction models are publicly available to facilitate future transcriptome mapping studies in diverse populations. See our 2018 article in PLOS Genetics for more information.
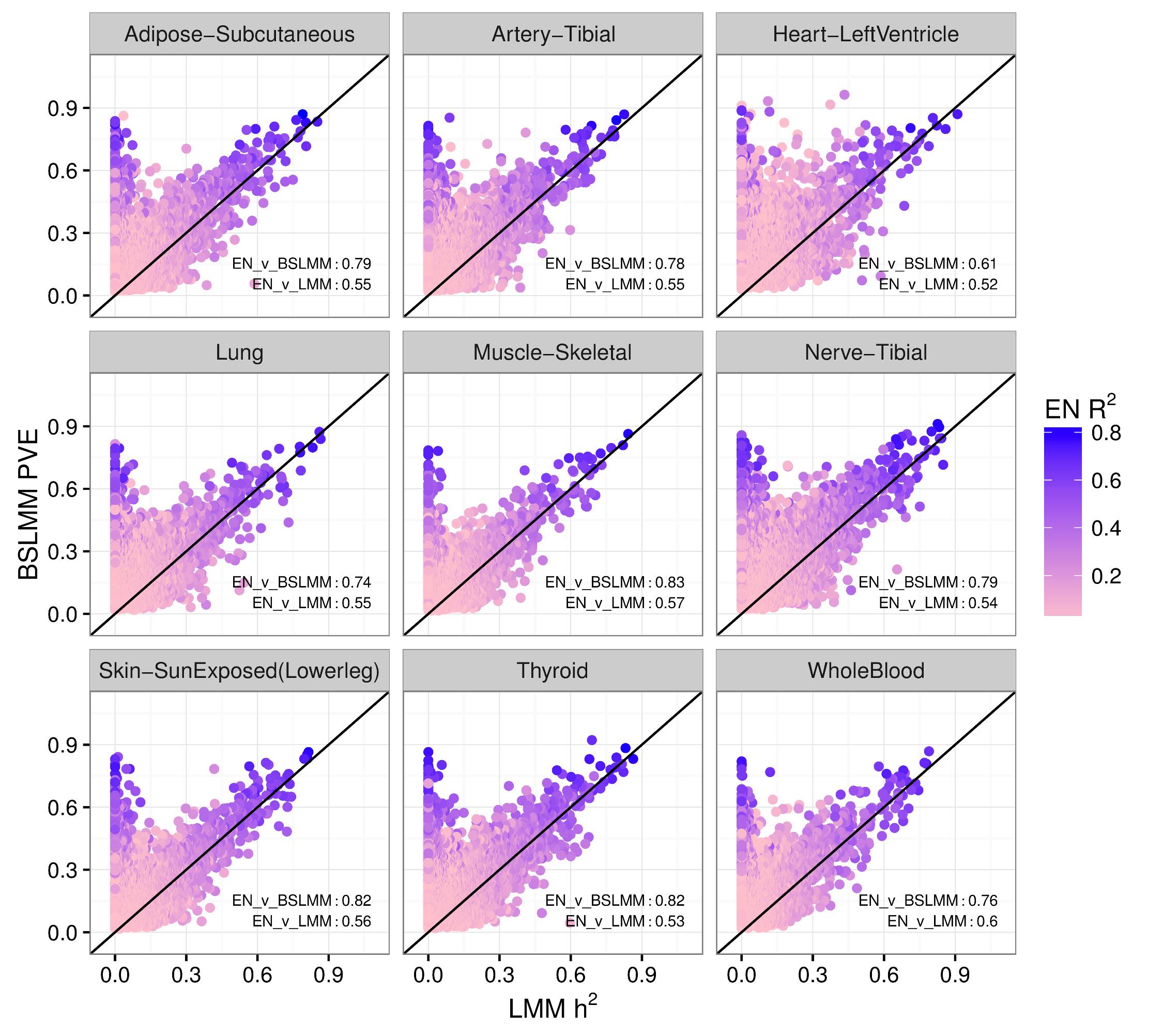
We performed a systematic survey of the heritability and the distribution of effect sizes across all representative tissues in the human body. Bayesian Sparse Linear Mixed Model (BSLMM) analysis provides strong evidence that the genetic contribution to local expression traits is dominated by a handful of genetic variants rather than by the collective contribution of a large number of variants each of modest size. In other words, the local architecture of gene expression traits is sparse rather than polygenic across all 40 tissues (from DGN and GTEx) examined. For most genes BSLMM PVE (percent variance explained) estimates are larger than LMM estimates reflecting the fact that BSLMM yields better estimates of heritability because of its ability to account for the sparse component. This result is confirmed by the sparsity of optimal performing gene expression predictors via elastic net modeling. See our 2016 article in PLOS Genetics for more information.
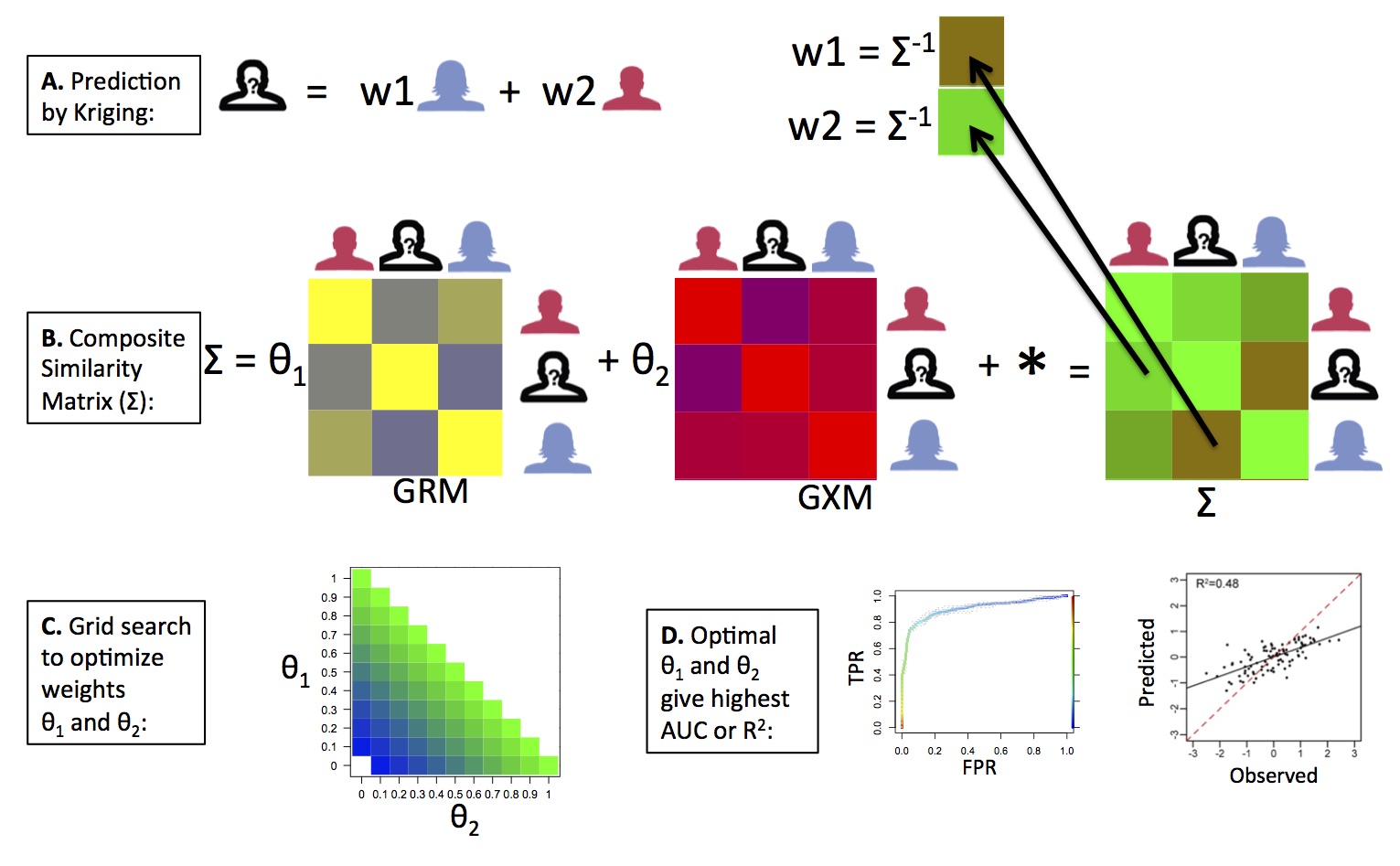
OmicKriging
We have developed a novel systems approach to complex trait prediction, which leverages and integrates similarity in genetic, transcriptomic or other omics-level data. Our method called OmicKriging emphasizes the use of a wide variety of systems-level data, such as those increasingly made available by comprehensive surveys of the genome, transcriptome and epigenome, for complex trait prediction. For example, integrating different omic correlation matrices such as a genetic relationship matrix (GRM) derived from SNPs and a gene expression correlation matrix (GXM) derived from gene expression levels may optimize predictive performance over either dataset alone. See our article in Genetic Epidemiology for more details.
Funding and Collaborations
NIH R15 HG009569 05/01/17 – 07/31/26
Predicting Gene Regulation Across Populations to Understand Mechanisms Underlying Complex Traits
Our project will lead to a better understanding of the effect and impact of genetic variation on gene expression and complex traits across populations, which has the potential to improve the implementation of precision medicine among diverse populations and reduce health disparities.
Role: PI
Loyola Strategic Plan 2020 Health-EQ Award 08/01/17 – 06/30/18
‘Omics integration in populations of African ancestry to understand mechanisms underlying obesity and cardiovascular disease
Our project will integrate DNA and RNA data with additional phenotype information (obesity, CVD risk) in an African-origin cohort to better understand biological mechanisms contributing to these comorbidities.
Role: Co-PI with Lara Dugas
NIH R01 MH107666 08/21/15 – 06/30/18
Predicted Gene Expression: High Power, Mechanism, and Direction of Effect
The goal of this project is to develop and apply methods that fully harness gene regulation within complex psychiatric trait association and prediction studies.
Role: Co-investigator (PI: Hae Kyung Im)
NIH R01 CA157823 09/25/12 – 07/31/17
Genetic Susceptibility and Biomarkers of Platinum-Related Toxicities
The goal of this project is to understand the biologic/genetic basis of acute/late chemotherapy-related toxicity in testicular cancer patients treated with cisplatin.
Role: Co-investigator (PI: Lois Travis)